CRISPR-Cas9/Cas12A expression, purification and validation
 In brief
In brief
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated) technologies enable scientists to accurately modify the genome of cells and organisms. CRISPR-Cas9 and CRISPR-Cas12a enzymes are the most widely studied of the RNA-guided endonucleases used for genome editing. To help reduce costs of our genome editing research projects, we have established an "in-house" protocol for purifiying and functionally testing a range of Cas9/Cas12a endonuclease which are attached to various affinity tags.
Highlights
- "In-house" purified CRISPR-Cas9 and CRISPR-Cas12a endonuclease can help to reduce laboratory costs of genome editing research.
- "In-house" purified CRISPR-Cas9 and CRISPR-Cas12a endonuclease offers flexibility to research labs who may want to produce uniquely modified/tagged proteins which are not commercially available.
- "In-house" purified Cas9 and Cas12a show comparable quality with commercial available products.
Introduction
Genome editing can change an organism’s genetic information by inserting, replacing or deleting a certain part of DNA sequence. The CRISPR-Cas technology allows the genetic editing with a single base-pair precision. It uses a small piece of RNA to guide a Cas endonuclease to the complementary region of the genome. The Cas endonuclease cuts the DNA, breaking both strands of its double helix. Naturally, cells will start to repair the damaged DNA using another similar piece of intact DNA as a template, which is known as homology-directed repair (HDR). Scientists utilize the HDR mechanism to introduce an extra piece of repair template DNA carrying desired mutations (insertion/deletion/replacement), and precisely edit the genes. However, mammalian cells rarely use the template DNA to repair the damage. Instead, mammals tend to fix double-stranded breaks in DNA by simply joining the broken ends together, which is called Non-Homologous End-Joining (NHEJ) and is prone to errors.
To minimize the NHEJ, scientists have started to investigate physically linking the repair template DNA to the Cas endonuclease, which increases the repair template DNA concentration in the nuclei and facilitates the HDR pathway (Savic, Ringnalda et al., 2018). A fusion tag on the Cas endonuclease forms a covalent bond between the endonuclease and the donor repair template, which enhances the high-fidelity repair due to an increase of donor template concentration in the nucleus region.
We have purified and tested a range of these modified CRISPR-Cas variants, in which DNA repair template is covalently conjugated to Cas endonuclease. This protocol outlines the method we use to express the CRISPR-Cas endonuclease proteins in E. coli. The proteins are purified by affinity chromatography and size exclusion chromatography following extraction from the cell lysate. This is followed by an in vitro activity test and quality control.
Materials & equipment
Materials
- Plasmid (for Cas9/Cas12a expression or activity test) and target sgRNA (IDT or Synthego)
- Antibiotics (ampicillin, kanamycin, or chloramphenicol etc.)
- Nickel-charged IMAC resin
- SDS-PAGE pre-casting gel
- NEBuffer and Cas9/Cas12a reaction buffer
- Proteinase K
- Tris-Glycine running buffer
- LB broth
- PBS
- Tris, HEPES, NaCl, KCl, imidazole
- Agarose gel
- IPTG
- Coomassie Blue, methanol, acetic acid
- Protein sample buffer (glycerol, SDS, β-mercaptoethanol, 0.5 M Tris pH 6.8 and bromophenol blue)
- PierceTM LAL chromogenic endotoxin quantitation kit
Equipment
- FPLC (AKTA system)
- Size exclusion column (e.g. Superdex 200 Increase 10/300 GL)
- Ion exchange column (e.g. HiTrap column SP HP)
- High-speed centrifuge
- Incubator/shaker with temperature controller
- Spectrophotometer or Nanodrop®
- PowerPacTM power system (Bio-Rad)
- Probe sonicator (or cell disrupter)
Procedure
Protein Expression
- Inoculate 10 ml of culture medium containing appropriate antibiotics (i.e. 100 ug/ml ampicillin, 50 ug/ml kanamycin, 34 ug/ml chloramphenicol or combinations) and grow an overnight culture with shaking at 37oC.
- Inoculate 800 mL of prewarmed media (with antibiotics) with 10ml of the overnight culture and grow at 37oC with vigorous shaking until an OD600 of 0.6 - 0.8 is reached.
- Take a 1 ml sample immediately before induction as a negative control (-IPTG).
- Induce expression by adding isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5-1 mM.
- Incubate the culture for an additional 1-5h at 37oC or overnight at 22 oC (depending on the expression level of different proteins, some proteins are not able to reach high expression without long induction times).
- Lower temperature is necessary to keep some proteins soluble and prevent formation of inclusion bodies.
- Harvest the cells by centrifugation at 4000 x g for 30 min at room temperature or 4 oC.
- Cell pellet can be stored at -20 oC or subject to different experiments.
Protein Purification
- Thaw the cell pellet on ice and resuspend the cells in lysis buffer (20 mM HEPES, 200 mM KCl, 20mM imidazole, pH 7.5) at 2 - 5 ml per gram wet weight cells. Keep a small sample at each step, to assist with trouble-shooting.
- Sonicate on ice using a probe sonicator or cell disrupter.
- Centrifuge lysate at 20,000 x g for 30 min at 4 ºC to pellet the cellular debris. Save the supernatant.
- Add 1 ml of the 50% w/v Ni2+-NTA slurry to 5 ml cleared lysate and mix gently by shaking (200 rpm on a rotary shaker) at 4ºC for 1 hour or overnight.
- Load the lysate and Ni2+-NTA mixture into a column with the bottom outlet capped.
- Remove bottom cap and collect the column flow-through.
- Wash column twice with 4 ml wash buffer (20 mM HEPES, 200 mM KCl, 50 mM imidazole, pH7.5); collect wash fractions for SDS-PAGE analysis.
- Add 1.5 ml elution buffer (20 mM HEPES, 200 mM KCl, 400 mM imidazole, pH7.5) to column and collect the eluate in tubes. Collect 10 column volumes of the elution fractions.
- (Optional) Following the Ni2+-NTA affinity chromatography, the sample can be also applied to size exclusion chromatography or ion-exchange chromatography connected with a FPLC system (GE Healthcare, AKTA) for further purification.
- Purge/wash (10 mL/min) the system (including all line system and fraction collector dispensor) with 20% ethanol, then water and buffer (0.22-μm protein-compatible filtered and degassed).
- Mount the column vertically on a stable stand, switch the flow speed to column default speed (such as 0.5 mL/min), and connect the column from pump with an extension to the FPLC system (connect top, then bottom). Adjust the pressure alarms (pre-column, ∆column, and system pressure)
- Equilibrate the column with buffer for more than one column volumn (CV)
- Inject the sample (maximum 1 mL sample for Superdex 200 increase 10/300GL column)
- Collect the sample from fraction collector and concentrating.
- Analyse each elution fraction by SDS-PAGE.
- The tank is filled with 1× Tris-Glycine running buffer (25 mM Tris pH8.3, 192mM Glycine, 0.1%SDS,) and the gels are run on the gels bench at 100 V for 1.5 hours.
- Following electrophoresis, separate gel from the glass plates and carefully transfer to a clean container containing the staining solution (0.25g Coomassie Blue R250, 40% methanol, 10% glacial acetic acid in distilled water and filtered through Whatman filter paper).
- Incubate at room temperature for 1 hour with gentle agitation on rocking platform.
- Pour the staining solution into a waste bottle (can be reused) and rinse gel with distilled water.
- Add destaining solution (40% methanol, 10% acetic acid in distilled water) and incubate at room temperature.
- Change the destaining solution three times until background levels are satisfactory.
- Rinse gel well in distilled water to stop further destaining.
- Dry gel onto Whatman filter paper or between cellophane sheets for future reference.
Validation of Cas9/Cas12a in vitro activity
- Prepare double stranded substrate DNA by PCR or double digestion from a plasmid of your choice.
- Assemble the reaction at room temperature in the following order:
20 ul Cas9 in-vitro reaction
H2O X 10× cas9/cas12a reaction buffer 2 2uM sgRNA 0.45 1uM Cas9 (NEB, PCV, MAV) or Cas12a 1 TOTAL (ul) 20 - Incubate mixture at 25 °C for 10 minutes
- Add substrate DNA (~350 ng) in the mixture and mix thoroughly.
- Incubate at 37°C for 30 minutes (can extend upto 60 minutes).
- Add 1 μl of Proteinase K (20 mg/ml) to each sample, Mix thoroughly and spin in a microfuge.
- Incubate at room temperature for 10 minutes.
- Proceed with gel analysis
Endotoxin level test (microplate assay procedure)
- Since the proteins are required for in-cell assay, an endotoxin assay needs to be done for quality control.
- Equilibrate all reagents to room temperature and pre-equilibrate the microplate in a heating block for 10 minutes at 37 °C before use. Maintainthe the microplate at 37 °C.
- Samples were diluted 1/10 and standards were 1/2 diluted.
- Dispense 50µL of each standard or sample replicate into the microplate well. The blank contains 50µL of endotoxin-free water.
- At time T=0, add 50µL of LAL reagent to each well using a pipettor. Begin timing as the LAL is added. Once the LALhas been added into all plate wells, briefly remove from the heating block and gently tap several times to facilitate mixing. Cover the plate with the lid and return to heating block to incubate at 37°C °C for 10 minutes.
- After 10 minutes (T=10), add 100µL of chromogenic substrate solution (prewarmed to 37 °C) to each well. Once the substrate solution has been added into all plate wells, briefly remove from the heating block and gently tap several times to facilitate mixing. Cover the plate with lid and return to heating block to incubate the plate at 37°C for 6 minutes.
- At T=16 minutes, add 100 µL of stop reagent (i.e. 25% acetic acid). Once the stop reagent was added into all plate wells, remove the plate from heating block and gently tap several times to facilitate mixing.
- Measure the absorbance at 405-410nm on a plate reader.
- Subtract the average absorbance of the blank replicates from the average absorbance of all individual standards and sample replicates to calculate mean ∆ absorbance.
- Prepare a standard curve by plotting the average blank-corrected absorbance for each standard on the y-axis vs. the corresponding endotoxin concentration in EU/mL on the x-axis. The coefficient of determination, r2, must be ≥0.98.
- Use the formula for a standard curve (linear regression) to determine the endotoxin concentration of each sample.
Worked example
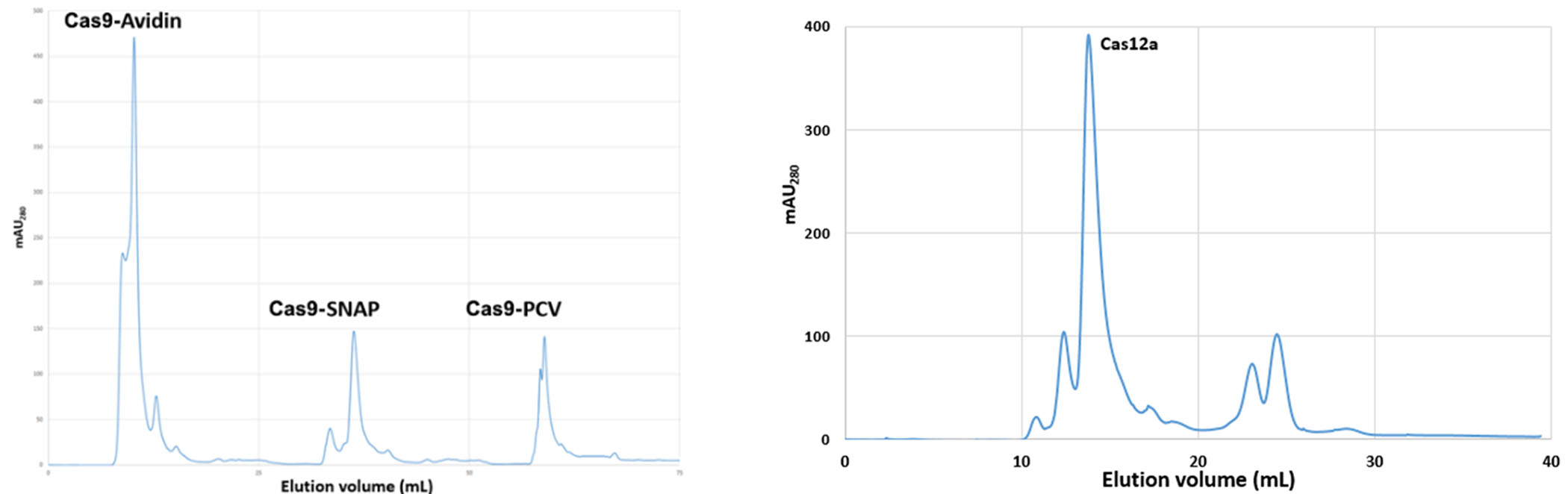
Cas9/Cas12a expression and affinity purification

Size exclusion purification
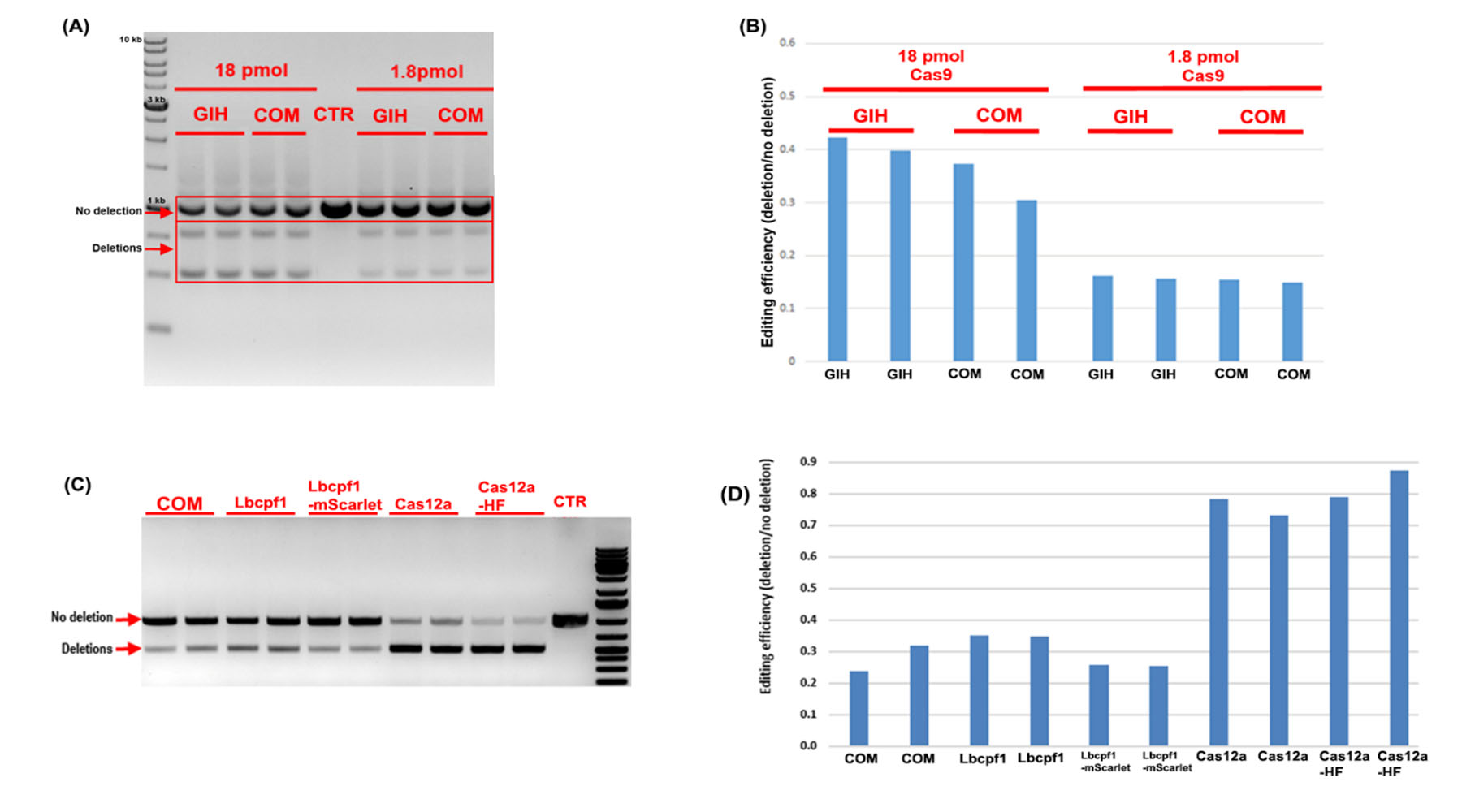
GIH in-house Cas9/Cas12a activity test comparing to commercial Cas9/Cas12a activity with two concentrations, i.e. 18 pmol v.s. 1.8 pmol
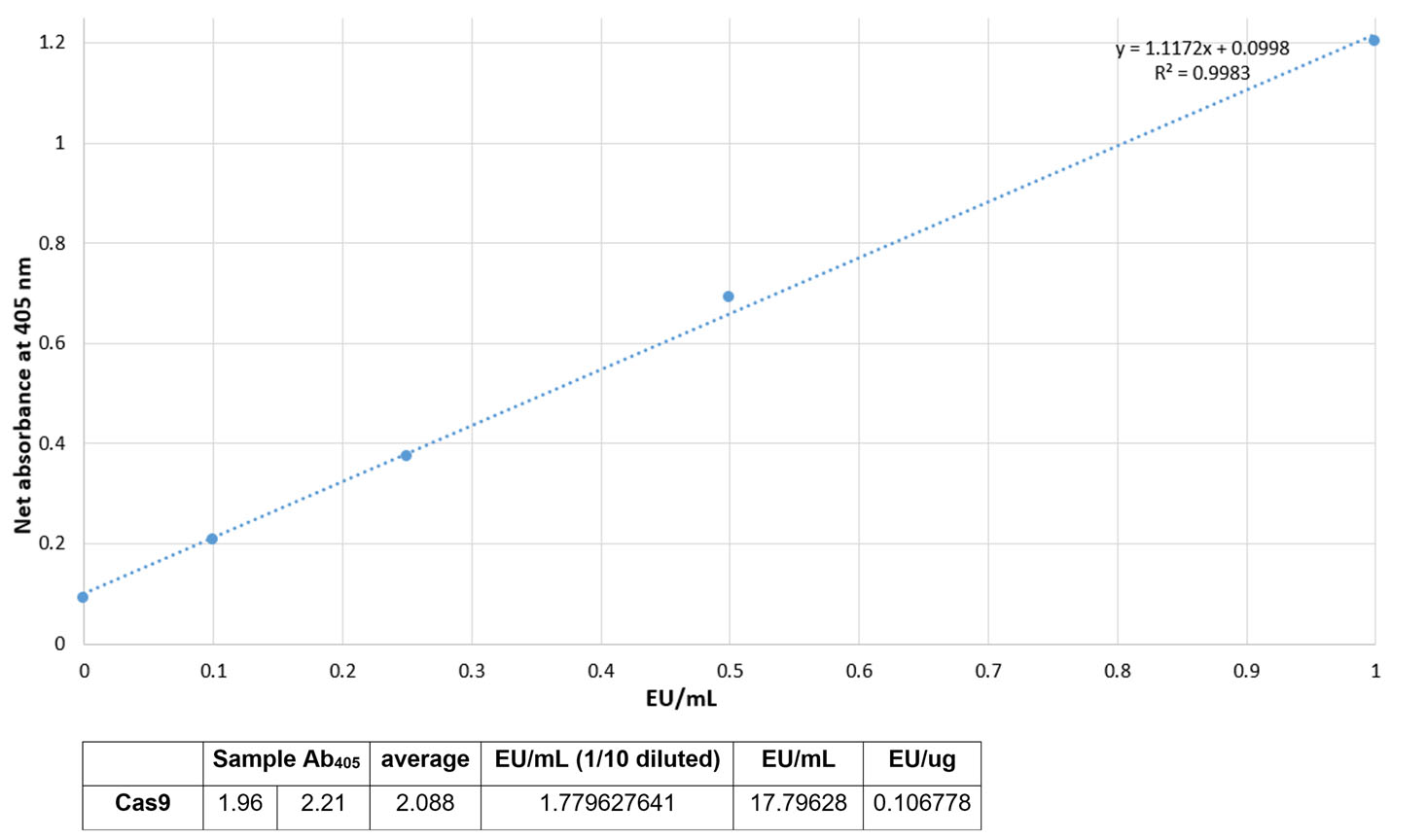
Endotoxin level test
Trouble-shooting
- Do not add glycerol during elution step, because its viscosity reduces the buffer exchange dramatically
- Work at low temperature environment during the process because the Cas9 is temperature sensitive
- Bacteria is cultured at low temperature (16-18 °C) to help with protein folding and solubility.
- Avoid the pressures over the pressure limit during FPLC
- Clean-in-place can be done after the columns are used several times.
- Final protein product needs to be sterilized by passing though 0.22 uM PVDF filter before using in genome editing experiments in cells or in vivo to prevent contamination.
References
- Rajagopalan, N., et al. (2018). "A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies." Methods and Protocols 1(2): 17.
- New England Biolabs. In vitro digestion of DNA with Cas9 Nuclease, S. pyogenes (M0386), DOI: dx.doi.org/10.17504/protocols.io.idhca36
- Savic N, Ringnalda FC, Lindsay H, Berk C, Bargsten K, Li Y, Neri D, Robinson MD, Ciaudo C, Hall J, Jinek M, Schwank G. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife. 2018 May 29;7:e33761. doi: 10.7554/eLife.33761. PMID: 29809142; PMCID: PMC6023611.
Associated project
-
Precise genome editing
January 2019
Standard operating procedure (SOP)
Please contact Stacey Andersen (operations manager) for the updated SOP.
GIH team


Collaborators
