Successful project outcomes
The GIH projects showcased here represent collaborations with evidence of direct research outcomes of benefit to the broader research community. They cover a broad range of sample types, technologies and outputs, celebrating the diverse areas GIH supports with the research undertaken at UQ.
GIH is proud to have contributed to these researchers sucessess and encourage you to seek out more information about any developments and outcomes of interest. Please note, the majority of these research outcomes are ongoing and we encourage you to check back over time for further updates.
Assembly of complex genome (Macadamia) using single molecule sequencing (2019)
Prof Robert Henry (Queensland Alliance for Agriculture and Food Innovation (QAAFI)), Prof Lachlan Coin, Dr Agnelo Furtado (QAAFI), Dr Tim Bruxner (Institute for Molecular Bioscience (IMB)), Joanna Crawford (IMB).
Spearheaded by our GIH collaborative project, the australian wild macadamia has provided a sophisticated model for the assembly of complex plant genomes.

The rapid advancement of DNA sequencing technologies has enabled the assembly of highly accurate, haplotype-resolved plant genomes at the chromosome scale. However, selecting the most suitable and cost-effective method from the range of options available can prove challenging for researchers, as sequencing complex plant genomes is a considerably long and expensive proposition.
Now published in the journal GigaScience, we undertook a GIH collaborative project with a team led by Professor Robert Henry at The Queensland Alliance for Agriculture and Food Innovation (QAAFI) and with additional investment from Hort Innovation’s Tree Genomics project, the Department of Agriculture and Fisheries Australia and Industry partner BGI Genomics Company. Our aim was to investigate the latest technology to improve the efficiency of this process for the benefit of all researchers.
We benchmarked 3 long-read sequencing technologies applied to the assembly of the genome Australian wild Macadamia, Macadamia jansenii, then QAAFI researchers assembled all 14 chromosomes of the species. Critically endangered, there are only around 100 trees of Macadamia jansenii left in the wild; all trees are growing in the same area near Miriam Vale in Queensland. Conservation efforts to protect the trees have welcomed our collaborative genomic research.
Optimised through the course of this project is a plant DNA extraction method, which attracted significant global attention as one of our most viewed social media posts with more than 5000 impressions on our UQ GIH Twitter feed. Our research data contributed to a follow-up publication in Gigabyte, presented in two webinars, hosted and recorded by BGI genomics and Australian BioCommons as well as a presentation by GIH at a major international genomics conference and several news and media articles.
The macadamia data we have generated has been fed through to a range of projects including research on sustainably intensifying tree crop production and breeding for key commercial attributes in macadamia production” said Professor Robert Henry.
Since our initial study, the three other Australian macadamia species, which are also on a Threatened Species list, have undergone the same analysis that the Genome Innovation Hub and collaborators spearheaded.
See the GIH project page for up-to-date details of outcomes of this project here.
Precise genome editing (2019)
Dr Nathan Palpant, Institute for Molecular Bioscience (IMB)
Our optimised protocol for purification of CRISPR-Cas9 and CRISPR-Cas12a endonuclease helps to reduce costs and offers flexibility to produce non- commercially available proteins of high quality in standard research laboratories.
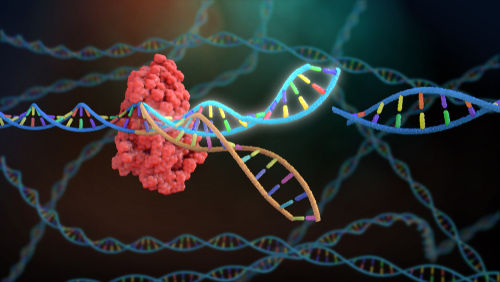 Genome editing can change an organism’s genetic information by inserting, replacing or deleting a certain part of DNA sequence. The CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated) technology allows the genetic editing with a single base-pair precision. It uses a small piece of RNA to guide a Cas endonuclease to the complementary region of the genome. The Cas endonuclease cuts the DNA, breaking both strands of its double helix. Naturally, cells will start to repair the damaged DNA using another similar piece of intact DNA as a template, which is known as homology-directed repair (HDR). Scientists utilize the HDR mechanism to introduce an extra piece of repair template DNA carrying desired mutations (insertion/deletion/replacement), and precisely edit the genes. However, mammalian cells rarely use the template DNA to repair the damage. Instead, mammals tend to fix double-stranded breaks in DNA by simply joining the broken ends together, which is called Non-Homologous End-Joining (NHEJ) and is prone to errors. To minimize the NHEJ, scientists have started to investigate physically linking the repair template DNA to the Cas endonuclease, which increases the repair template DNA concentration in the nuclei and facilitates the HDR pathway (Savic, Ringnalda et al., 2018). A fusion tag on the Cas endonuclease forms a covalent bond between the endonuclease and the donor repair template, which enhances the high-fidelity repair due to an increase of donor template concentration in the nucleus region.
Genome editing can change an organism’s genetic information by inserting, replacing or deleting a certain part of DNA sequence. The CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated) technology allows the genetic editing with a single base-pair precision. It uses a small piece of RNA to guide a Cas endonuclease to the complementary region of the genome. The Cas endonuclease cuts the DNA, breaking both strands of its double helix. Naturally, cells will start to repair the damaged DNA using another similar piece of intact DNA as a template, which is known as homology-directed repair (HDR). Scientists utilize the HDR mechanism to introduce an extra piece of repair template DNA carrying desired mutations (insertion/deletion/replacement), and precisely edit the genes. However, mammalian cells rarely use the template DNA to repair the damage. Instead, mammals tend to fix double-stranded breaks in DNA by simply joining the broken ends together, which is called Non-Homologous End-Joining (NHEJ) and is prone to errors. To minimize the NHEJ, scientists have started to investigate physically linking the repair template DNA to the Cas endonuclease, which increases the repair template DNA concentration in the nuclei and facilitates the HDR pathway (Savic, Ringnalda et al., 2018). A fusion tag on the Cas endonuclease forms a covalent bond between the endonuclease and the donor repair template, which enhances the high-fidelity repair due to an increase of donor template concentration in the nucleus region.
In this GIH collaborative project with the research group of Dr Nathan Palpant, from the Institute for Molecular Bioscience, our aim was to purify and test a range of these modified CRISPR-Cas variants, in which DNA repair template is covalently conjugated to Cas endonuclease. Consequently, we have established an "in-house" protocol for purifiying and functionally testing a range of Cas9/Cas12a endonuclease which are attached to various affinity tags. Our "In-house" protocol for purification of CRISPR-Cas9 and CRISPR-Cas12a endonuclease reduces costs (compared to commercial products) and offers flexibility to research labs who may want to produce uniquely modified/tagged proteins (which are not commercially available). Most importantly, our "In-house" purified Cas9 and Cas12a show comparable quality with commercial available products. Our purified Cas proteins have been successfully used in a number of GIH projects and by collaborative researchers.
The GIH optimised protocol, including anticpated results, activity testing and trouble shooting tips is available for all researchers via the GIH website.
See the GIH project page for up-to-date details of outcomes of this project here.
Spatial genomics technologies to study cancer and genetic diseases in tissue contexts (2019)
Dr Quan Nguyen, A/Prof Andrew Mallett, Institute for Molecular Bioscience (IMB).
Revolutionising insight into cancer through gene expression analysis of cells in whole tissues.
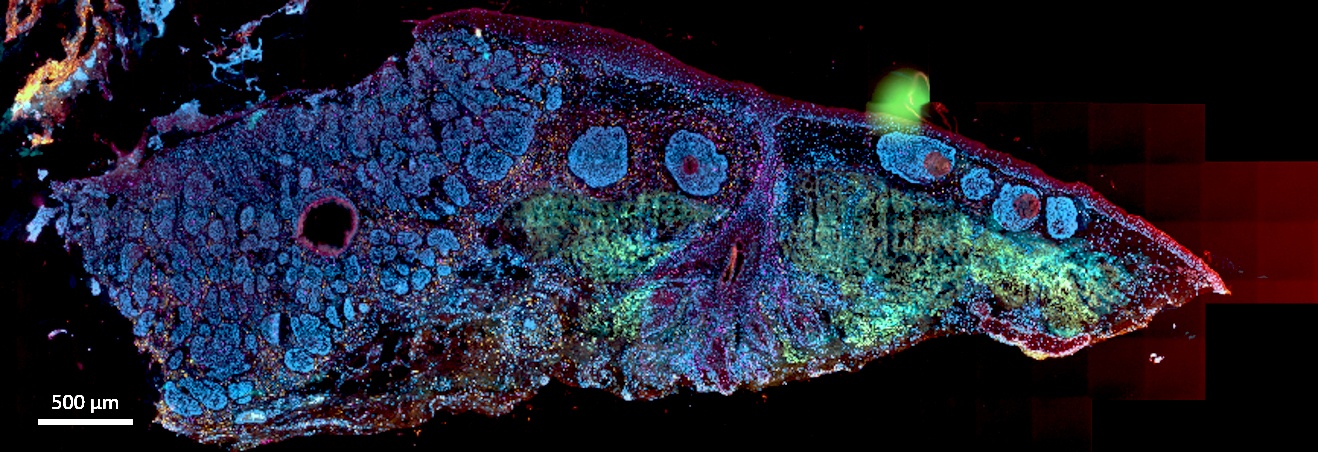 Crowned as the "Method of the Year 2020" by Nature Methods, spatially resolved transcriptomics, which is the ability to measure gene activity where it occurs, or "spatially", in a whole tissue sense, has revolutionised how researchers can investigate biology and disease. Achieved by a combination of high-resolution sequencing and imaging data on whole tissue sections, it offers revolutionary insight into complex cellular interactions where they occur.
Crowned as the "Method of the Year 2020" by Nature Methods, spatially resolved transcriptomics, which is the ability to measure gene activity where it occurs, or "spatially", in a whole tissue sense, has revolutionised how researchers can investigate biology and disease. Achieved by a combination of high-resolution sequencing and imaging data on whole tissue sections, it offers revolutionary insight into complex cellular interactions where they occur.
In this GIH collaborative project with the research groups of Dr Quan Nguyen and Prof Andrew Mallett, we have developed Spatial transcriptomics analysis and machine learning pipelines using the technologies Visium (10X Genomics) and RNAscope (ACD Biosciences).
“The expert help from GIH staff has been instrumental for my group to successfully set up some of the pioneering genomics technologies in Australia. Our spatial transcriptomics platform comprises of imaging and sequencing technologies as well as bioinformatics analysis. GIH staff helped us achieving all three components. This platform now is available to all researchers at UQ, with multiple collaborative projects with different schools and institutes. The collaborations have resulted in publications and successful grant funding.” Dr Quan Nguyen, Senior Research Fellow -GL, Institute for Molecular Bioscience.
Published in the journal Bioinformatics, as well as available as open source-software, the user-friendly deep learning software, SpaCell, was initially developed to analyse gene expression measurements from spatially barcoded spots in a tissue. This was followed up by the development of two other bioinformatic tools; stLearn - to comprehensively analyse Spatial Transcriptomics (ST) data to investigate complex biological processes within an undissociated tissue and STRISH - a computational pipeline that enables us to quantitatively model cell-cell interactions by automatically scanning for local expression of RNAscope data to recapitulate an interaction landscape across the whole tissue and, as well as three further manuscripts under review for publication highlighting the use of these analysis software in skin breast and brain cancer as well as the pathology of chronic kidney disease.
These innovations in the ability to utilise spatial omics technology in exploring how individual cells exist within their tissue microenvironment, though transcriptome analysis is showing great promise in areas such as cancer pathology. Traditional histopathological analysis of biopsies is time-consuming and can be highly variable, which is an important consideration for diagnoses in cancers such as brain tumour and early melanoma.The long-term goal of computer aided diagnosis is to improve accuracy and speed to beyond that possible as seen with the human eye. Additionally, understanding the genomics within tumour sites could potentially identify treatment options within critical timeframes.
Easily accessible by UQ researchers, are the whole spatial transcriptomics pipelines we have developed, from the GIH-optimised technical protocols available via the GIH website, to full integration into UQ infrastructure sequencing and imaging facilities, as well as the free-to-use analysis software.
“I have found the depth of technical expertise and personal assistance from the GIH team pivotal to the success of my PhD research in molecular profiling of human kidney cancer. Without their support it would not have been feasible to apply cutting edge molecular techniques in limited and irreplaceable clinical cancer tissue. In addition, their continued support has helped me build a strong collaborative partnership between diagnostic, clinical and research domains between UQ-IMB, Pathology Queensland, Princess Alexandra Hospital, Royal Brisbane and Women’s Hospital, Translational Research Institute and Queensland Institute of Medical Research.” Arti Mala Raghubar, PhD candidiate, Faculty of Medicine
See the GIH project page for up-to-date details of outcomes of this project here.
Dual gRNA screen (2019)
 Cell states are governed by complex heterotypic interactions between regulatory genes. Previous work has used genetic loss of function to study the complex interdependent roles of transcription factors controlling gene expression, differentiation, and morphogenesis. In this GIH collaborative project with the research group of Dr Nathan Palpant, from the Institute for Molecular Bioscience, we have developed an industry partnership with Agilent to establish a scalable screening platform using standard CRISPR editing strategies.
Cell states are governed by complex heterotypic interactions between regulatory genes. Previous work has used genetic loss of function to study the complex interdependent roles of transcription factors controlling gene expression, differentiation, and morphogenesis. In this GIH collaborative project with the research group of Dr Nathan Palpant, from the Institute for Molecular Bioscience, we have developed an industry partnership with Agilent to establish a scalable screening platform using standard CRISPR editing strategies.
While previous studies have been limited to analysis of 1-3 regulatory genes in a combinatorial manner, in this project we aimed to utilize scalable bar-coded RNA-seq with a library of multiplexed dual-gRNA gene knockouts to systematically map the heterotypic roles and interdependencies of transcription factors and epigenetic regulators controlling cell identity. As such, we have generated a workflow for high efficient CRISPR knockout using optimised multiple guide RNAs design. In association with the Queensland Facility for Advanced Genome Editing (QFAGE) and the Transgenic Animal Service of Queensland (TASQ), this workflow has now contributed to two recent excellent publications by UQ researchers. Published in Circulation, by Dr Meredith Redd and colleagues as "Therapeutic Inhibition of Acid Sensing Ion Channel 1a Recovers Heart Function After Ischemia-Reperfusion Injury", their data provides compelling evidence for a novel pharmacologic strategy involving ASIC1a blockade as a cardioprotective therapy to improve the viability of hearts subjected to Ischemia–reperfusion injury . GIH's Dr Di Xia used this workflow for the CRISPR transfection, troubleshooting and analysis required to generate the ASIC1a knock-out iPSCs utilised in the publication.
In a second publication, released in the Journal of Cell Biology, Dr Harriet P. Lo and colleages as 'Cavin4 interacts with Bin1 to promote T-tubule formation and stability in developing skeletal muscle', they identified a role for the muscle-specific component, Cavin4, in skeletal muscle T-tubule development by analyzing two vertebrate systems, mouse and zebrafish. In this research, Dr Di Xia utilised the CRISPR workflow for both the genome editing required to create both the Cavin4 Knockout mouse model and Cavin4 Knockout C2C12 cells.
See the GIH project page for up-to-date details of outcomes of this project here.
Quantitative transcriptome analysis (CAGE) (2019)
 CAGE (cap analysis gene expression) is highly sensitive and accurate quantitative transcriptome analysis which utilizes a cap structure on 5’ end of RNA. This feature enables high-throughput identification of transcription start sites and ability to infer promoters and transcription factor binding motifs. This project established the availability of CAGE through development of an optimized, cost -efficient in-house protocol, benchmarked against a commercially available ‘gold standard’ kit. his developed "In-house" CAGE protocol delivers high quality results at a ~1/3 of the reagent cost and 25% less hands-on time when compared to the commercial kit.. CAGE can be used to simultaneously locate transcription start sites and quantify the abundance of RNA transcripts.
CAGE (cap analysis gene expression) is highly sensitive and accurate quantitative transcriptome analysis which utilizes a cap structure on 5’ end of RNA. This feature enables high-throughput identification of transcription start sites and ability to infer promoters and transcription factor binding motifs. This project established the availability of CAGE through development of an optimized, cost -efficient in-house protocol, benchmarked against a commercially available ‘gold standard’ kit. his developed "In-house" CAGE protocol delivers high quality results at a ~1/3 of the reagent cost and 25% less hands-on time when compared to the commercial kit.. CAGE can be used to simultaneously locate transcription start sites and quantify the abundance of RNA transcripts.
A full detailed protocol including anticipated results and troubleshooting is available from the GIH website. To our knowledge this was the first successful implementation of CAGE in Australia. Several research groups have utilised the CAGE protocol for generating data within their own areas of interest.
See the GIH project page for up-to-date details of outcomes of this project here.
Genome-wide CRISPR screening for modifiers of diverse cellular phenotypes (2019)
Our optimised a pipeline for pooled genome-wide CRISPR knockout and activation screens can be used to identify modifiers of diverse cellular phenotypes.
CRISPR/Cas9 technology, which was first used for gene editing in 2013, has revolutionised our ability to genetically manipulate cells and organisms, and represents one of the most exciting advances in cellular biology in the 21st century due to the diversity of its potential applications. Co-expression of Cas9, an RNA-guided DNA endonuclease, and a synthetically designed single guide ribonucleic acid (sgRNA) in cells can accurately target any gene of interest for CRISPR gene knockout (CRISPR-ko) or activation (CRISPR-a).
The recent availability of well-characterised genome-wide CRISPR-ko and CRISPR-a sgRNA libraries represents a powerful new resource. Pooled genome-wide libraries consist of DNA constructs that have the same backbone but code for different sgRNAs, meaning that CRISPR/Cas9 machinery can be directed to every protein-encoding gene in the human genome for genome-wide knockout or activation.
 In this project driven by Dr Rebecca San Gil and Dr Adam Walker, from the Queensland Brain Institute, we successfully optimised protocols to conduct pooled human genome-wide CRISPR knockout and activation screens with FACS-based, high-throughput screening of cell phenotypes in established cell lines. Genomic DNA from FACS-purifed samples were extracted and underwent next generation sequencing. Sequencing data was subsequently analysed by GIH bioinformatician Mr Jon Xu, using multiple different algorithms. This analysis revealed, in a non-biased approach, gene targets and biological processes that protected against or sensitised cells to toxicity in the model systems utilised.
In this project driven by Dr Rebecca San Gil and Dr Adam Walker, from the Queensland Brain Institute, we successfully optimised protocols to conduct pooled human genome-wide CRISPR knockout and activation screens with FACS-based, high-throughput screening of cell phenotypes in established cell lines. Genomic DNA from FACS-purifed samples were extracted and underwent next generation sequencing. Sequencing data was subsequently analysed by GIH bioinformatician Mr Jon Xu, using multiple different algorithms. This analysis revealed, in a non-biased approach, gene targets and biological processes that protected against or sensitised cells to toxicity in the model systems utilised.
“I was the recipient of a 2019 and 2021 GIH external project grant/collaboration to develop and expand on CRISPR-based technologies at the University of Queensland. This opportunity has been outstanding for me as an early career researcher a number of reasons; it was a chance to prepare and submit a project grant application as lead investigator, I was able to propose and successfully deliver a technically challenging project to a high standard through collaboration with GIH staff, GIH has helped to establish me as a CRISPR-expert at UQ, which has led to grant success and collaborations with other UQ researchers.” Dr Rebecca San Gil, Fight MND Early Career Research Fellow
Targetting 20,000 genes of the human genome in a single experiment is a powerful functional genomics tool for application to diverse biological questions at UQ. We welcome opportunities for further collaboration using this technique!
See the GIH project page for up-to-date details of outcomes of this project here.
TraDIS-Vault: an interactive searchable genome browser and repository for TraDIS data (2020)
Prof Ian Hendserson, Dr. Dom Gorse, Institute for Molecular Biosciences (IMB) and QCIF Bioinformatics.
TraDIS-vault is a flexible and user-friendly, free online viewer and web-based public interface for storage, easy visualisation and comparison of hundreds of TraDIS bacterial data sets.
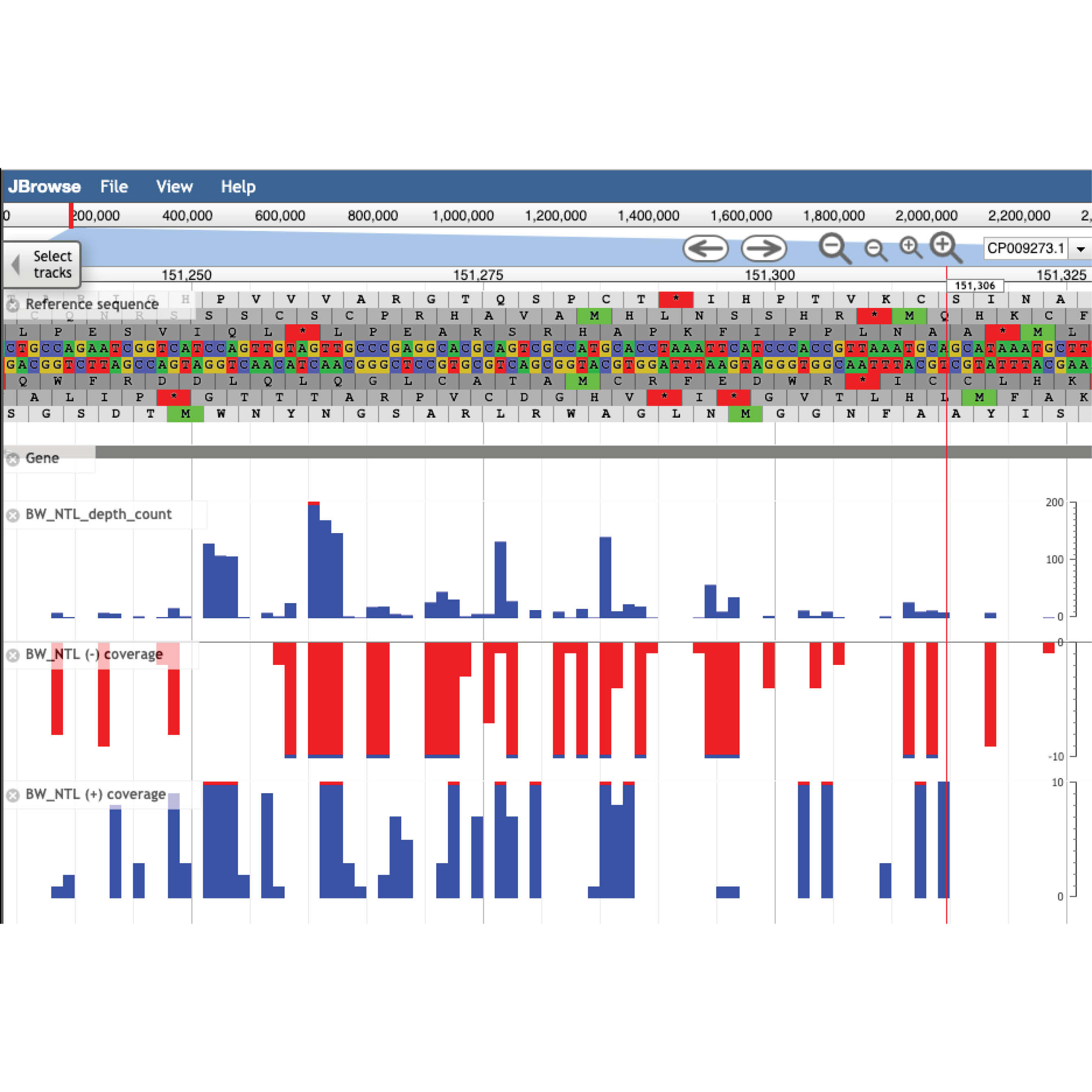 TraDIS, or Transposon Directed Insertion Site Sequencing, is a genome sequencing technique involving inserting transposons (‘jumping genes’) into a genome to generate large numbers of mutants. TraDIS-vault is based on Web Apollo, a platform for genome annotation that is widely adopted by the genomics community. It is currently the only resource of its kind and was mentioned in a recent Nature review.
TraDIS, or Transposon Directed Insertion Site Sequencing, is a genome sequencing technique involving inserting transposons (‘jumping genes’) into a genome to generate large numbers of mutants. TraDIS-vault is based on Web Apollo, a platform for genome annotation that is widely adopted by the genomics community. It is currently the only resource of its kind and was mentioned in a recent Nature review.
Four members of the QCIF Bioinformatics team collaborated with GIH on the TraDIS-vault project led by Professor Ian Henderson, Director of UQ’s Institute for Molecular Bioscience. The Henderson research group studies bacteria and their interaction with humans and animals. TraDIS is the approach they use to identify genes encoding essential functions. The Henderson group’s current TraDIS library is the densest mutant library described for any organism, positioning the group uniquely within the global community. QCIF Computational Biologist Michael Thang, who worked closely with the Henderson group, said the project team used QRIScloud, QCIF’s cloud compute service, to host a customised Web Apollo server.
“We’ve tailored the navigation and visualisation of the genome browser to make the rich information contained in TraDIS-vault more accessible to the research community. Researchers need more and more interactive tools to expose their research data and QCIF is well set up to help,” said Michael.
The project team will expand TraDIS-vault to assimilate relevant microbial RNAseq and ChIPseq data sets into the database, providing a comprehensive tool for analysing and integrating genetic information. The genome browser is not intended for hosting raw data and private tracks, but data already published and publicly available. Display up to 32 tracks, explore by organism, condition or genetic interactions and export your chosen region of interest.
See the GIH project page for up-to-date details of outcomes of this project here.
Simultaneous identification of RNA-chromatin interactions and transcriptomes in single cells (2020)
 Epigenetic heterogeneity in cancer can drive tumour evolution and adaptive drug resistance. Current approaches for understanding the mechanisms of action of chromatin-associated RNAs are limited by low sensitivity. UQ researchers Dr Seth Cheetham and Dr Adam Ewing from the Mater Research Institute, Faculty of Medicine with Professor Geoff Faulkner from the Queensland Brain Institute, have previously demonstrated that lncRNA-chromatin interactions can be identified at ultra-high sensitivity by tethering the adenine methylase Dam to MS2-tagged chromatin-associated lncRNAs in Drosophila. Recently, the transcriptomes and chromatin-occupancy of Dam fusion proteins were simultaneously determined in single cells. In collaboration with GIH, the aim was to further this research and develop a protocol for single-cell identification of RNA-chromatin associations and their transcriptomic effects in single mammalian cells. This has led to the pre-print Single-molecule simultaneous profiling of DNA methylation and DNA-protein interactions with Nanopore-DamID and GIH support acknowledged in a a further two pre-prints Long-read cDNA sequencing identifies functional pseudogenes in the human transcriptome and Methylartist: Tools for Visualising Modified Bases from Nanopore Sequence Data, which includes development of the computational tool "Methylartist", freely available on github and which has also been highlighted directly on the Oxford Nanopore website.
Epigenetic heterogeneity in cancer can drive tumour evolution and adaptive drug resistance. Current approaches for understanding the mechanisms of action of chromatin-associated RNAs are limited by low sensitivity. UQ researchers Dr Seth Cheetham and Dr Adam Ewing from the Mater Research Institute, Faculty of Medicine with Professor Geoff Faulkner from the Queensland Brain Institute, have previously demonstrated that lncRNA-chromatin interactions can be identified at ultra-high sensitivity by tethering the adenine methylase Dam to MS2-tagged chromatin-associated lncRNAs in Drosophila. Recently, the transcriptomes and chromatin-occupancy of Dam fusion proteins were simultaneously determined in single cells. In collaboration with GIH, the aim was to further this research and develop a protocol for single-cell identification of RNA-chromatin associations and their transcriptomic effects in single mammalian cells. This has led to the pre-print Single-molecule simultaneous profiling of DNA methylation and DNA-protein interactions with Nanopore-DamID and GIH support acknowledged in a a further two pre-prints Long-read cDNA sequencing identifies functional pseudogenes in the human transcriptome and Methylartist: Tools for Visualising Modified Bases from Nanopore Sequence Data, which includes development of the computational tool "Methylartist", freely available on github and which has also been highlighted directly on the Oxford Nanopore website.
See the GIH project page for up-to-date details of outcomes of this project here.
Developing a scalable genome browser and repository for complex multi-omic datasets (2021).
 Genomic, transcriptomic, proteomic and associated approaches are being employed in increasingly wider contexts to understand biological systems and how they change. This necessarily means moving beyond a handful of well-supported model organisms to diverse wild, non-model organisms that are driving an avalanche of ‘omics data being generated across the globe. Unfortunately, datasets are often fragmented and intractable, and thus of limited utility beyond the initial study. This huge untapped potential can be released by making data much more accessible and usable – i.e. democratised – by integrating all datasets related to a specific genome into a comprehensive, visual, standardised and publicly-available system.
Genomic, transcriptomic, proteomic and associated approaches are being employed in increasingly wider contexts to understand biological systems and how they change. This necessarily means moving beyond a handful of well-supported model organisms to diverse wild, non-model organisms that are driving an avalanche of ‘omics data being generated across the globe. Unfortunately, datasets are often fragmented and intractable, and thus of limited utility beyond the initial study. This huge untapped potential can be released by making data much more accessible and usable – i.e. democratised – by integrating all datasets related to a specific genome into a comprehensive, visual, standardised and publicly-available system.
To realise this potential, with Professors Sandie and Bernard Degnan from UQ's Degnan Marine Genomics Labs, within the School of Biological Sciences, Faculty of Science and in collaboration with QCIF Bioinformatics we have developed an expandable Apollo browser as an interactive repository for diverse transcriptomic, chromatin-state and proteomic data.
“The Degnan Marine Genomics Lab (School of Biological Sciences) is very excited to be seeing the realisation of their new scalable genome browser and interactive data repository, currently being developed as a 2021 GIH collaborative project with QCIF Bioinformatics and Australian Biocommons. The system will transform the way in which the enormous body of data that has been generated by the Degnan group can be explored, analysed and shared with collaborators across the world. Importantly, outcomes from the project will be of enormous value far beyond the Degnan Lab, as the finished platform will be made available to all research groups looking for a cleverly-designed browser that allows users to search, visualise, interrogate and share their ‘omics data on non-model organisms.”
Existing genomes of two Great Barrier Reef animals are being deployed to populate and test the browser as it's being developed: (i) the reef flat demosponge Amphimedon queenslandica and its proteobacterial symbionts; and (ii) the notorious destroyer of coral reefs, the crown-of-thorns starfish (COTS). Existing ‘omics datasets for these species are then be mapped to the genomes, able to be visualised as customised on-demand/collapsible tracks. The success of this project has lead to multiple news and media releases as well as a webinar launching the new Apollo Service hosted by Australian Biocommons (now viewable on youtube).
See the GIH project page for up-to-date details of outcomes of this project here.