Full title
Genotype-free demultiplexing of pooled single-cell RNA-seq.
Genome Biol 20, 290 (2019). https://doi.org/10.1186/s13059-019-1852-7
https://doi.org/10.1186/s13059-019-1852-7
About
"Splitting" mixed samples in RNA sequencing: reducing costs, increasing accuracy in droplet-based scRNA-seq.
Using single-cell RNA sequencing (scRNA-seq) to cell biology at cellular level provides greater resolution than “bulk” level analyses, thus allowing more refined understanding of cellular heterogeneity. For example, it can be used to cluster cells into sub-populations based on their differential gene expression, so that different fates of cells during development can be discovered. Droplet-based scRNA-seq (for example Drop-Seq [1] or 10X Genomics Systems [2]) allows profiling large numbers of cells for sequencing by dispersing liquid droplets in a continuous oil phase [3]s in an automated microfluidics system, and as a result is currently the most popular approach to scRNA-seq despite a high cost per run. Methods that lower the per sample cost of running scRNA-seq are required in order to scale this approach up to a population scale. An effective method for lowering scRNA-seq cost is to pool samples prior to droplet-based barcoding with subsequent demultiplexing of sequence reads.
Experimental approaches
Cell hashing [4] based on Cite-seq [5] is one such experimental approach to demultiplex pooled samples. This approach uses oligo-tagged antibodies to label cells prior to mixing, but use of these antibodies increases both the cost and sample preparation time per run. Moreover, it requires access to universal antibodies for organism of interest, thus limiting applicability at this stage to human and mouse. Alternatively, computational tools like demuxlet [6] have been developed to demultiplex cells from multiple individuals, although this requires additional genotyping information to assign individual cells back to their samples of origin. This limits the utility of demuxlet, as genotype data might not be available for different species; biological material may not be available to extract DNA; or the genetic differences between samples might be somatic in origin.
Another issue for droplet-based scRNA-seq protocols is the presence of doublets, which occurs when two cells are encapsulated in same droplet and acquire the same barcode. The proportion of doublets increases with increasing number of cells barcoded in a run. It is imperative that these are flagged and removed prior to downstream analysis. Demuxlet [6] uses external genotype information to address this issue, and other tools have been developed to solve this issue based on expression data alone, including Scrublet [7] and Doubletfinder [8].
Our bioinformatic tool
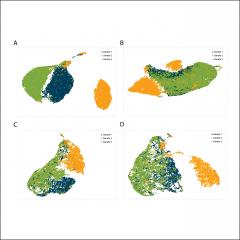
Here we introduce a simple, accurate, and efficient tool, mainly for droplet-based scRNA-seq, called "scSplit", which uses a hidden state model approach to demultiplex individual samples from mixed scRNA-seq data with high accuracy. Our approach does not require genotype information from the individual samples to demultiplex them, which also makes it suitable for applications where genotypes are unavailable or difficult to obtain. scSplit uses existing bioinformatics tools to identify putative variant sites from scRNA-seq data, then models the allelic counts to assign cells to clusters using an expectation-maximisation framework.
Further resources
More detailed information about scSplit can be found in both the publication and on our development page
GIH contribution

This publication is the sucessful outcome of the GIH development "scSplit tool: genotype-free demultiplexing of pooled single-cell RNA-seq". Primary author and GIH team member Jun Xu designed the algorithms, implemented the tools in Python and tested it on multiple datasets.
Jun Xu also drafted the manuscript for publication.
Follow his twitter account here: @xujun_jon

Contributing author and team member of both GIH and the IMB Sequencing Facilty, Stacey Andersen, participated in important discussions and provided useful suggestions on multiple issues.